

Sterben In ISO 14155: 2020 Ist ein Standard mit dem Titel, der bisher nicht für den MDR harmonisiert wurde „Klinische Untersuchungen von medizinischen Geräten für menschliche Probanden – gute klinische Praxis“. Auf Deutsch: “Klinische Untersuchung von medizinischen Geräten – gute klinische Praxis”.
Es beschreibt somit den Stand der Technik für Hersteller von Medizinprodukten bei der Vorbereitung, Planung, Implementierung und Bewertung klinischer Tests und bestimmt die Verantwortung der beteiligten Akteure, insbesondere des sogenannten Sponsors.
Dieser Artikel hilft bei einem Überblick über ISO 14155 und mit Tipps zur Verwendung des Standards, um die regulatorischen Anforderungen für klinische Tests so schnell und ungehindert wie möglich zu erfüllen.
1. ISO 14155: Eine Einführung
1.1 Kontext
Der MDR hat die Anforderungen an klinische Daten erhöht, mit denen Hersteller von Medizinprodukten die Leistung, Sicherheit und Wirksamkeit ihrer Produkte nachweisen müssen. Daher sind mehr Hersteller verpflichtet, diese Daten im Kontext klinischer Studien, in bestimmten klinischen Untersuchungen, zu sammeln. Der MDR stellt auch umfangreiche Anforderungen für diese klinischen Untersuchungen.
ISO 14155 sollte den Herstellern in Zukunft in Zukunft in Zukunft bedienen.
1.2 Anwendungsbereich von ISO 14155
Einerseits gilt der Standard für klinische Tests von medizinischen Geräten, die dem Ziel dienen, die Leistung, Sicherheit und Wirksamkeit der Produkte zu demonstrieren. Andererseits fallen auch die anderen „klinischen Studien nach der Platzierung auf den Markt“ (nach dem Wortlaut des Standards) in den Geltungsbereich von ISO 14155. Dies sind beispielsweise nach dem Markt für klinische Follow-up-Untersuchungen (PMCF).
1,3 Ziele der Norm
Der Standard verfolgt mehrere Ziele:
- Schutz der Subjekte (Sicherheit, Brunnen, Rechte)
- Resilienz der Ergebnisse
- Klarheit über die Verantwortlichkeiten, insbesondere der Sponsor (z. B. Hersteller), Haupttester und potenzielle andere Teilnehmer (z. B. CRO, Krankenhaus)
- Uniformes Verständnis aller Teilnehmer (Sponsoren, Prüfer, Ethikkommissionen, Behörden, benannte Körperschaften)
1.4 Verwendung für die Hersteller
Daher profitieren die Hersteller von Medizinprodukten vom Standard:
- Weniger regulatorische Unsicherheit und Diskussion mit Behörden und ernannten Behörden
- Infolgedessen schneller und weniger komplexe “Zustimmung” der Produkte
- Sparen Sie Zeit durch konkrete Richtlinien und Beispiele, z. B. zur Struktur von Dokumenten wie dem klinischen Testplan und dem klinischen Testbericht
2. Struktur von ISO 14155: 2020
Der Standard umfasst über hundert Seiten und besteht aus zehn Kapiteln und zehn Anhängen (siehe Abb. 2).
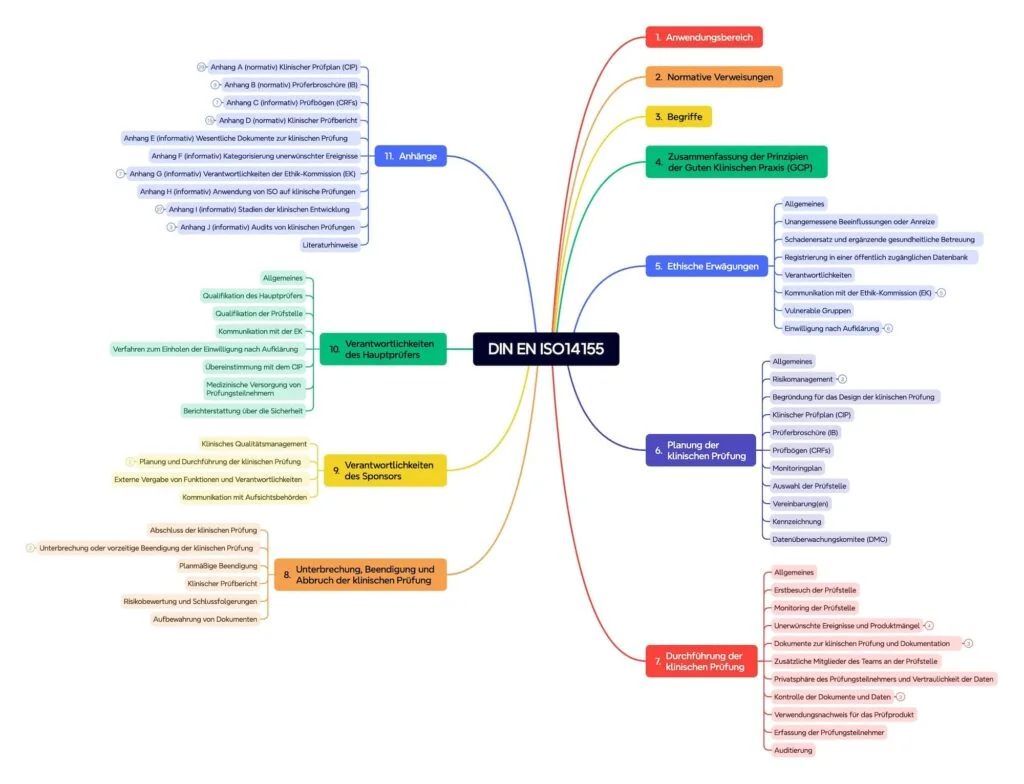
Kapitel 4: Gute klinische Praxis
Kapitel 4 fasst die Prinzipien der guten klinischen Praxis (GCP) zusammen.
Kapitel 5: Ethische Überlegungen
Kapitel 5 enthält ethische Überlegungen. Es untersucht die Kommunikation mit der Ethikkommission, der Verantwortlichkeiten, der gefährdeten Gruppen, der Einwilligungserklärung und der Patienteninformationen.
Kapitel 6: Planung der klinischen Untersuchung
Kapitel 6 beschreibt die Planung des klinischen Tests. Dazu gehören:
- Bestimmungen zum Testprodukt
- Spezifikationen für das Design der klinischen Untersuchung
- Spezifikationen zur Auswahl des Testzentrums und der wesentlichen Dokumente der klinischen Untersuchung
Diese Dokumente umfassen:
- Klinischer Testplan (CIP – – Klinischer Untersuchungsplan)
- Testbroschüre (ib – – Ermittler Broschüre)
- Bögen testen (CRFs – – Case-Report-Formen)
Die Anhänge liefern weitere Informationen zur Einrichtung dieser Dokumente.
Kapitel 7: Umsetzung der klinischen Untersuchung
Kapitel 7 nennt die Anforderungen für die Implementierung des klinischen Tests:
- Informationen zur praktischen Implementierung
- Nachrichten von Ereignissen oder Produktfehlern
- Umgang mit Dokumenten und Daten
- Implementierung der Überwachung
- Vertraulichkeit der Daten und Identifizierung der Prüfungsteilnehmer
Kapitel 8: Unterbrechung, Beendigung und Abriss der klinischen Untersuchung
Hier sind die Anforderungen an Unterbrechung, Beendigung und Abbruch der klinischen Untersuchung enthalten. Dies betrifft auch den klinischen Testbericht, die Risikobewertung und die Speicherung der Dokumente.
Kapitel 9 und 10: Verantwortung
Kapitel 9 und 10 Regulieren Sie die Verantwortlichkeiten des Sponsors und des Hauptprüfers. Die Anforderungen an den Sponsor sind für Hersteller von Medizinprodukten besonders relevant.
Normative und informative Anhänge
Die umfangreichen Anhänge der ISO 14155 sind manchmal normativ.
- Klinischer Testplan (CIP) (normativ): definierter Inhalt des CIP
- Dozentbroschüre (IB) (normativ): Mindestanforderungen für den Inhalt des IB
- Testbögen (CRFs) (informativ): Tipps zum Erstellen von CRFs
- Klinischer Testbericht (normativ): Definierter Inhalt des klinischen Testberichts am Ende der klinischen Untersuchung
- Wesentliche Dokumente für klinische Tests (informativ): Dokumente, die vor, während und nach der klinischen Untersuchung im Sponsor oder im Testzentrum verfügbar sein müssen
- Kategorisierung unerwünschter Ereignisse (informativ): Übersichtstabelle und Grafiken zur Orientierung
- Verantwortlichkeiten der Ethikkommission (EK) (informativ): Leitfaden zur guten Praxis in Ethikkommissionen
- Anwendung von ISO 14971 auf klinische Prüfungen (informativ): Grafiken für den Umgang mit Risiken während einer klinischen Untersuchung
- Stadien der klinischen Entwicklung (informativ): Überblick über die verschiedenen Entwicklungsarten klinische Untersuchungen des Medizinprodukts und die Anwendbarkeit von ISO 14155 auf die verschiedenen Arten
- Audits von klinischen Prüfungen (informativ): Tipps, wie der Sponsor nachweisen kann, dass die klinische Untersuchung im Einklang mit GCP stattfindet
3. Wichtige Anforderungen von ISO 14155
3.1 Ethische Überlegungen
Klinische Untersuchungen müssen gemäß den ethischen Grundsätzen durchgeführt werden. Der ISO 14155 muss immer mit der aktuellen Version der Deklaration von Helsinki verwendet werden, in der die ethischen Grundsätze für die Durchführung klinischer Prüfungen definiert werden.
Vorhersehbare Risiken müssen gegen den erwarteten Nutzen abgewogen werden.
Die Rechte, die Sicherheit und das Bohrloch der Teilnehmer haben vor wissenschaftlichen Interessen Priorität.
3.2 bedingungslose wissenschaftliche Strenge
Die Prüfung muss klare und verständliche statistische Anforderungen folgen, die mit hohen Beweisen gut begründete Ergebnisse bringen.
Präklinische und klinische Informationen müssen den Test ausreichend unterstützen.
3.3 umfassende Dokumentation
Eine gründliche und verständliche Dokumentation aller Phasen des klinischen Tests ist erforderlich.
Die Umsetzung muss gemäß den von der Ethikkommission und der Behörde genehmigten Dokumenten durchgeführt werden (UA -Testplan, Handbuch des klinischen Prüfers, Patienteninformationen).
3.4 Klare Verantwortlichkeiten
Klare Definitionen der Rollen und Verantwortlichkeiten aller Beteiligten sind unerlässlich und müssen schriftlich festgelegt werden.
4. Stolpersteine, die vermieden werden müssen
4.1 Entdeckte Bedingungen beim Hersteller
Die Annahme, dass der Hersteller die Rolle des Sponsors im klinischen Test leicht übernehmen kann, kann häufig angenommen werden.
Das Sponsoring im Rahmen einer klinischen Studie ist mit einigen Spezifikationen verbunden, einschließlich der Integration des klinischen Qualitätsmanagements in die vorhandenen QMs des Herstellers.
Denken Sie also daran, dass Sie die Prüfung nur starten können, wenn Sie als Sponsor die Anforderungen des Standards und des MDR erfüllen.
4.2 Fehler bei der Anwendbarkeit von ISO 14155
“Wir machen nur eine PMCF -Studie, das Produkt ist bereits zertifiziert, daher gilt die Norm nicht.”
Dies ist ein Missverständnis.
Die Norm gilt nicht nur für “Zulassungsstudien”, die durchgeführt werden, um das CE zu erhalten. Es muss auch in Teilen für Studien zur Klärung wissenschaftlicher Fragen (Artikel 82 nach MDR) oder in PMCF -Studien (Artikel 74 Studien) verwendet werden. Dies wird in Anhang I.7 des Standards erläutert.
4.3 Fehlende Dokumentation
In der Regel reicht die Zusammenfassung der Studienergebnisse nicht aus, um die klinischen Beweise zu beweisen.
Hersteller müssen die klinischen Daten ihres eigenen Produkts aus Studien im Rahmen der klinischen Bewertung bewerten. Wenn Sie eine Studie mit Ihrem eigenen Produkt durchführen, wird erwartet, dass Sie die Studie der Studie vorlegen können, um die klinischen Beweise zu beweisen und die Konformität der Studien mit der ISO 14155 nachzuweisen. Dies wird vom genannten Körper als Teil der “klinischen Bewertung bewertet” (Abschnitt E des MDCG 2020-13) überprüft.
Im schlimmsten Fall könnten Zweifel an der Qualität der Daten entstehen, da ein Hersteller nicht ohne den Testplan und die anderen Dokumente beweisen kann, dass die Studie nach ISO 14155 durchgeführt wurde. Dies würde bedeuten, dass Sie die Daten gegebenenfalls ausschließen müssten.
4.4 Exklusiver Fokus auf ISO 14155
Es ist ein weit verbreiteter Fehler, dass die reine Einhaltung von ISO 14155 automatisch allen nationalen Anforderungen entspricht. Andererseits ist es richtig, dass nationale Anforderungen, insbesondere außerhalb der EU, berücksichtigt werden müssen.
5. Versionen von ISO 14155
5.1 Unterschiede zwischen den Versionen 2012 und 2020
ISO 14155: 2020-01 (oder DIN en ISO 14155: 2021-05) haben im Vergleich zur vorläufigen Form von 2012 mehrere bemerkenswerte Änderungen implementiert:
- Die Verflechtung mit der ICHP-GCP-Richtlinie wurde hervorgehoben und eine Zusammenfassung der GCP-Prinzipien in Abschnitt 4 in den Standard integriert.
- Das Risikomanagement wurde in den gesamten Prozess des klinischen Tests integriert.
- Der Anhang I, der für Hersteller besonders interessant ist, wurde eingeführt, was die Anwendbarkeit des Standards auf die verschiedenen klinischen Entwicklungsniveaus des medizinischen Geräts erläutert.
- Eine Anfrage zur Registrierung der klinischen Prüfung in einer öffentlich zugänglichen Datenbank wurde hinzugefügt (neu in Version 2020/2021).
- Das klinische Qualitätsmanagement (Abschnitt 9.1) und die risikobasierte Überwachung (Abschnitt 6.7) wurden integriert.
- Die Anpassung der internationalen Datenschutzanforderungen wurde in der aktuellen Version berücksichtigt.
5.2 Outlook für die nächste Version
Eine Änderung der ISO 14155 (DIN en ISO 14155/A11: 2024) wird derzeit geändert. Der Zeitpunkt der Veröffentlichung ist derzeit noch unbekannt (ab Juni 2025).
Die Überarbeitung zielt auch darauf ab, die klinischen Tests mit den EU -Anforderungen des MDR zu harmonisieren. Es sollte ohne Übergangsfrist gültig sein. (Quelle)
6. Schlussfolgerung und Zusammenfassung
ISO 14155 – Unabhängig von seiner Harmonisierung – ist der Goldstandard für die Vorbereitung, Planung, Implementierung und Bewertung klinischer Studien von medizinischen Geräten. Es gilt nicht nur für klinische Untersuchungen und sollte auch in Studien außerhalb Europas befolgt werden.
Der Standard ist ausreichend präzise und vollständig und daher die Hersteller mehr Vorteile als eine weitere behördliche Belastung.
Die „klinischen Experten“ des Johner -Instituts helfen Ihnen dabei, klinische Daten zu finden und zu entscheiden, ob eine klinische Studie überhaupt erforderlich ist.
In diesem Fall unterstützen wir Sie in Ihrer vollständigen klinischen Prüfung oder PMCF -Studie: Von der Planung bis zur Anwendung auf die Implementierung.
- Studiendesign: Wir helfen zu Design und Abstimmung.
- Statistische Fallnummernplanung: Optimale Planung für gültige Ergebnisse
- CRO-Auswahl: Wir finden die richtige Vertragsforschungsorganisation (CRO).
- Einreichung bei den Behörden: Schaffung von Regulierungsdokumenten
- Implementierung und Bewertung: Immer an Ihrer Seite, während Sie mit dem CRO im klinischen Bereich arbeiten
Hier finden Sie einen Überblick über die Unterstützung des Johner -Instituts für klinische Überprüfungen und Prüfungen.
Ähnliche Beiträge
Automotive
Game Center
Game News
Review Film
Berita Terkini
Berita Terkini
Berita Terkini
review anime

