
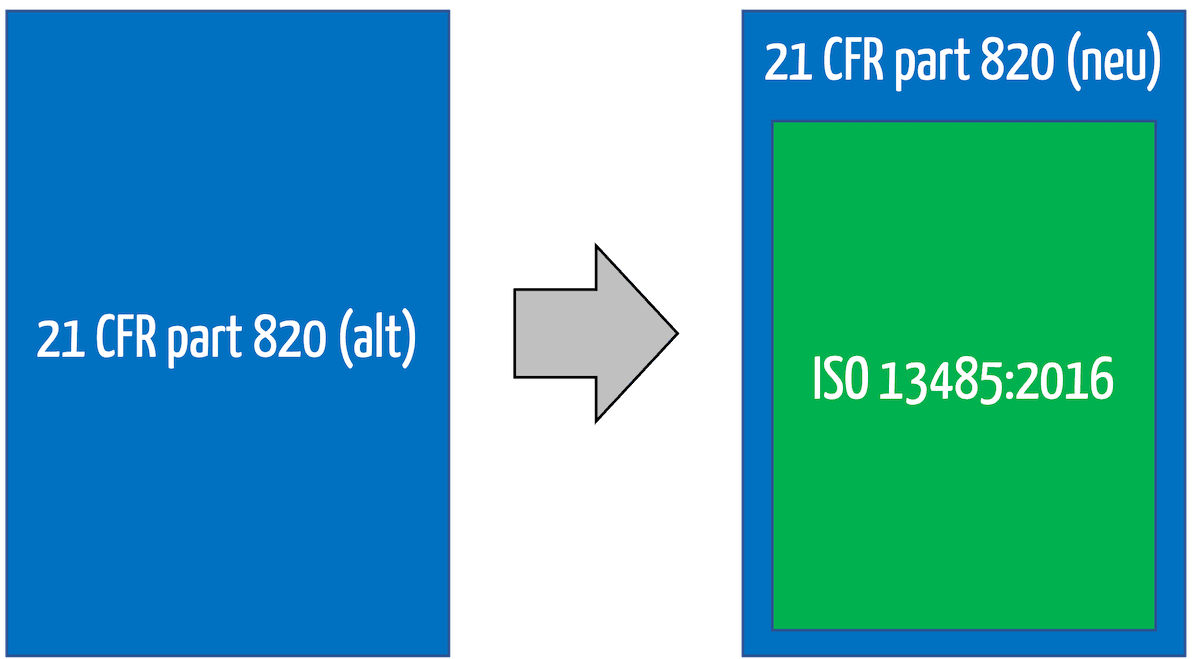
Ich bin 21 CFR Teil 820 Die FDA formuliert unter anderem die Anforderungen an die Qualitätsmanagementsysteme von Medizinprodukteherstellern. Dies bedeutet, dass 21 CFR Teil 820 (Qualitätssystemverordnung QSR) das Gegenstück zu ISO 13485.
1. QMSR: Die Änderungen an 21 CFR Teil 820
Denn die FDA hat am 2. Februar 2024 beschlossen, 21 CFR Part 820 weitgehend „auszuhöhlen“ und durch einen Verweis auf ISO 13485 zu ergänzen.
- Dieser Verweis befindet sich im neuen Abschnitt § 820.7 („Eingliederung durch Verweis“).
- Tabelle 1 zeigt, welche Abschnitte dadurch obsolet wurden.
Da ISO 13485 von Qualitätsmanagementsystemen spricht, hat die FDA beschlossen, den Titel von 21 CFR 820 umzubenennen. Dies wird nicht mehr Quality System Regulation (QSR) genannt. Vielmehr spricht die FDA nun von der Quality Management System Regulation (QMSR).
Mit dieser Änderung werden die bisherigen Anforderungen an eine Designhistoriendatei, einen Gerätestammsatz und einen Gerätehistoriensatz nicht mehr explizit erwähnt. Dennoch verlangt auch ISO 13485 ähnliche Aufzeichnungen.
a) Vergleich des alten und neuen 21 CFR Teil 820
Unterabschnitte AD
| 21 CFR Teil 820 alt (QSR) | 21 CFR 820 neu (QMSR) |
| Unterabschnitt A – Allgemeine Bestimmungen § 820.1 – Geltungsbereich. § 820.3 – Definitionen. § 820.5 – Qualitätssystem. |
Unterabschnitt A – Allgemeine Bestimmungen § 820.1 – Geltungsbereich. § 820.3 – Definitionen. § 820.7 – Einbeziehung durch Verweis. § 820.10 – Anforderungen an ein Qualitätsmanagementsystem. |
| Unterabschnitt B – Ergänzende Bestimmungen | |
| Unterabschnitt B – Anforderungen an das Qualitätssystem § 820.20 – Managementverantwortung. § 820.22 – Qualitätsaudit. § 820.25 – Personal. |
Gelöscht (reserviert für zukünftige Ergänzungen) |
| § 820.35 Kontrolle von Aufzeichnungen | |
| Unterabschnitt C – Designkontrollen § 820.30 – Designkontrollen. |
Gelöscht (reserviert für zukünftige Ergänzungen) |
| Unterabschnitt D – Dokumentenkontrollen § 820.40 – Dokumentenkontrollen. |
Gelöscht (reserviert für zukünftige Ergänzungen) |
Unterabschnitte EO
| 21 CFR Teil 820 alt | Geplante Änderungen |
| Unterabschnitt E – Einkaufskontrollen § 820.50 – Einkaufskontrollen. |
Gelöscht |
| Unterabschnitt F – Identifizierung und Rückverfolgbarkeit § 820.60 – Identifizierung. § 820.65 – Rückverfolgbarkeit. |
Gelöscht |
| Unterabschnitt G – Produktions- und Prozesskontrollen § 820.70 – Produktions- und Prozesskontrollen. § 820.72 – Inspektions-, Mess- und Prüfgeräte. § 820.75 – Prozessvalidierung. |
Gelöscht |
| Unterabschnitt H – Akzeptanzaktivitäten § 820.80 – Empfangs-, In-Process- und Fertiggeräteabnahme. § 820.86 – Akzeptanzstatus. |
Gelöscht |
| Unterabschnitt I – Nicht konformes Produkt § 820.90 – Nicht konformes Produkt. |
Gelöscht |
| Unterabschnitt J – Korrektur- und Vorbeugungsmaßnahmen § 820.100 – Korrektur- und Präventivmaßnahmen. |
Gelöscht |
| Unterabschnitt K – Kennzeichnung und Verpackungskontrolle § 820.120 – Gerätekennzeichnung. § 820.130 – Geräteverpackung. |
820.45 Gerätekennzeichnung und Verpackungskontrollen. |
| Unterabschnitt L – Handhabung, Lagerung, Vertrieb und Installation § 820.140 – Handhabung. § 820.150 – Lagerung. § 820.160 – Verteilung. § 820.170 – Installation. |
Gelöscht |
| Unterabschnitt M – Aufzeichnungen § 820.180 – Allgemeine Anforderungen. § 820.181 – Gerätestammdatensatz. § 820.184 – Aufzeichnung des Geräteverlaufs. § 820.186 – Aufzeichnung des Qualitätssystems. § 820.198 – Beschwerdeakten. |
Gelöscht Gelöscht § 820.35 Kontrolle von Aufzeichnungen (nur in Bezug auf UDI) Gelöscht § 820.35 Kontrolle von Aufzeichnungen |
| Unterabschnitt N – Wartung § 820.200 – Wartung. |
§ 820.35 Kontrolle von Aufzeichnungen (nur in Bezug auf Wartungsaufzeichnungen) |
| Unterabschnitt O – Statistische Techniken § 820.250 – Statistische Techniken. |
Gelöscht |
b) Unterschiede gegenüber ISO 13485
Die Qualitätsmanagementsystembestimmungen in Teil 820 und ISO 13485 sind nicht völlig identisch:
| Aspekt | Unterschiede |
| Umfang | Der Anwendungsbereich (§ 820.1) ist insgesamt unterschiedlich. Für Produkte der Klasse I (ausgenommen beispielsweise Produkte, die Software enthalten) verzichtet die FDA auf die Anforderungen von Kapitel 7.3 der ISO 13485 („Design und Entwicklung“). |
| Begriffsdefinitionen | Die FDA fügt in § 820.3 der ISO 13485 eigene Definitionen hinzu, beispielsweise „Komponente“, „fertiges Gerät“, „Wiederaufbereiter“. Sie definiert die Begriffe „Implantierbares medizinisches Gerät“, „Hersteller“, „Organisation“, „Rework“ und „Sicherheit und Leistung“ unterschiedlich. Dies bedeutet, dass die entsprechenden Begriffe in ISO 13485 überschrieben werden. |
| Produktidentifizierung, Rückverfolgbarkeit und Etikettierung | Hier ist die FDA konkreter und verlangt UDI gemäß Abschnitt 830 sowie „Rückverfolgbarkeit“ gemäß Abschnitt 821. Ebenso wie die MDR sind auch die Kennzeichnungsanforderungen spezifischer als die der ISO 13485. Sie finden sich im neuen § 820.45. |
| Wachsamkeit und Kommunikation mit Behörden | Für spezifischere Anforderungen verweist die FDA auf die Teile § 803 und § 806. |
| Dokumentation | Die FDA spezifiziert den Inhalt von Kundenreklamations- und Serviceaktivitätsaufzeichnungen genauer. |
Die Gründe und eine detaillierte Beschreibung der Änderungen finden Sie im Bundesregister. Dazu muss man sehr weit nach unten scrollen oder auf der Website nach dem Begriff „4. Überarbeiten Sie Teil 820, so dass er wie folgt lautet:“ suchen.
Eine Übersicht aller FDA-Anforderungen finden Sie hier unter dem Stichwort FDA.
c) Zeit- und Übergangsfristen
Die „endgültige Regelung“ für die QMSR wurde am 2. Februar 2024 veröffentlicht. Diese wird für einen Zeitraum von 2 Jahren, also am 2. Februar 2026, gültig und somit anwendbar sein. Bis dahin ist weiterhin die alte QMSR einzuhalten.
2. Der 21 CFR Teil 820 vor der Bezugnahme auf ISO 13485
a) Übersicht
Die Qualitätssystemvorschriften bestehen (vor der Bezugnahme) aus den Unterabschnitten A bis O, die die Abschnitte 1 bis 250 umfassen (siehe Tabelle 1 und Abbildung 2).
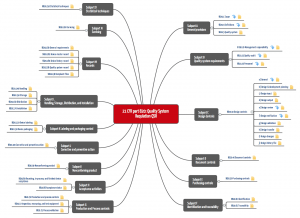
„Part 820“ fordert ein lückenloses Qualitätsmanagementsystem, das die Dokumentation und Umsetzung der „üblichen“ Verfahrensanweisungen vorschreibt. Dazu gehört:
- Dokumentenkontrolle
- Beschaffung
- Entwicklung
- Produktion
b) Anwendbarkeit von 21 CFR Teil 820
Abhängig von der Klasse des Medizinprodukts müssen Hersteller die allgemeinen Kontrollen des „Lebensmittel-, Arzneimittel- und Kosmetikgesetzes“ (§ 501 ff) und ab Klasse II auch die „besonderen Kontrollen“ einhalten. Die „General Controls“ enthalten bereits „Anforderungen an die gute Herstellungspraxis“, die sich auf die Entwicklung, Produktion, Verpackung, Lagerung und Installation der Geräte beziehen.
Genau diese Anforderungen an die „Current Good Manufacturing Practice (CGMP)“ sind Gegenstand der Qualitätssystemverordnung QSR. Dies bedeutet, dass diese Vorschriften von allen Medizinprodukteherstellern und auch anderen Akteuren wie Vertragsherstellern eingehalten werden müssen. Nur wenige Produkte der Klasse I sind ausgenommen bzw. GMP-frei. Die Einhaltung des CGMP prüft die FDA durch Inspektionen.
c) Entwicklungsanforderungen
Besonders relevant für die Entwicklungsabteilung ist der Bereich 820.30 mit den Design Controls. Dies gilt sogar für die Entwicklung von Medizinprodukten der Klasse I, wenn diese Software enthalten oder Software sind.
Die Anforderungen in 820.30 sind sehr allgemein. Aus diesem Grund orientieren sich beispielsweise auch Hersteller von Medizinprodukten, die Software enthalten, an den Guidance-Dokumenten der FDA. Diese beschreiben detailliert, wie die Designeingaben dokumentiert werden müssen.
Viele Hersteller unterschätzen die Bedeutung des Design History File DHF (820,30 j). Diese Dokumente müssen den Nachweis erbringen, dass die in 21 CFR Teil 820 beschriebenen Verfahren tatsächlich umgesetzt wurden und dass die Dokumentation nicht nachträglich erstellt wurde.
d) Unterschied zwischen 21 CFR 820 und ISO 13485
AnsonGroup hat einen Vergleich der Anforderungen von ISO 13485 und FDA QSR veröffentlicht. Einen weiteren Vergleich finden Sie hier. Die weitgehende Übereinstimmung zwischen den beiden Standards ist offensichtlich. Es sind jedoch Unterschiede zwischen ISO 13485 und Teil 820 zu beachten:
- Die Anforderungen an die Dokumentation sind höher; ISO 13485 erkennt auch nicht die logische Gruppierung von Dokumenten in die Design-History-Datei, den Gerätestammdatensatz und den Gerätehistoriendatensatz.
- Umgekehrt geht der Anspruch der ISO 13485 bzw. ISO 9001 an die Kundenzufriedenheit und die kontinuierliche Verbesserung des QM-Systems über die Anforderungen von QSR hinaus.
- Auch der Umgang mit Beschwerden und das Meldesystem unterscheiden sich deutlich.
Die FDA erkennt die ISO 13485-Zertifizierung nicht als Nachweis der Konformität mit den Anforderungen des 21 CFR Part 820 an. Im Gegensatz zur ISO 13485 gibt es auch keine Zertifizierung nach 21 CFR Part 820.
Bereits vor der Harmonisierung der QSRs mit ISO 13485 können Hersteller im Rahmen des Medical Device Single Audit Program MDSAP gleichzeitig die Konformität mit den Anforderungen von 21 CFR part 820 und ISO 13485:2016 nachweisen.

Der schnellste und kostengünstigste Weg zum Aufbau Ihres ISO 13485-konformen QMS
Mit unserer Videoschulung und Dutzenden Vorlagen in Auditgarant erhalten Sie eine umfassende Anleitung zum Aufbau eines schlanken und konformen Qualitätsmanagementsystems.
3. Fazit und Zusammenfassung
Es ist ein großer Fortschritt in der Harmonisierung regulatorischer Anforderungen, dass die FDA ihre Anforderungen an ein QM-System im 21 CFR Part 820 im Wesentlichen durch einen Verweis auf ISO 13485 ersetzt. Im „neuen“ 21 CFR Part 820 wird erläutert, wie bestimmte Anforderungen, beispielsweise zur Produktidentifizierung und Vigilanz, konkret zu erfüllen sind.
Für Medizinproduktehersteller wäre es hilfreich gewesen, wenn auch MDR und IVDR diesen Weg gegangen wären.
Benötigen Sie Unterstützung beim Aufbau oder der Verbesserung eines ISO 13485- oder/und FDA-konformen Qualitätsmanagementsystems? Oder benötigen Sie Hilfe beim Umstieg von QSR auf QMSR? Wir unterstützen Sie gerne!
Bei Bedarf übernehmen wir auch gerne die Rolle des US-Agenten.
Kontaktieren Sie uns zum Beispiel hier über das Kontaktformular.
Geschichte ändern
- 08.12.2025: Weiterer Vergleich verlinkt in 2.d).
- 21.02.2024: Überarbeitung auf Basis der verabschiedeten Fassung der QMSR vom 2. Februar 2024
Ähnliche Beiträge
Automotive
Agen Togel Terpercaya
Bandar Togel
Sabung Ayam Online
Berita Terkini
Artikel Terbaru
Berita Terbaru
Penerbangan
Berita Politik
Berita Politik
Software
Software Download
Download Aplikasi
Berita Terkini
News
Jasa PBN
Jasa Artikel


